If you haven't found answer to your question, please contact the Genomics Helpdesk to discuss any other 10x Genomics related problem.
We accept samples Monday to Thursday between 10am to 4pm (or 3pm if you submit nuclei for ATAC workflow). External users - when you arrive at the CRUK CI building please ask reception to call us and we will come to pick up samples from you.
We cannot accept samples that have not been discussed with our 10x team in advance (we usually ask for at least 1 weeks' notice). This is to ensure we have the reagents and time necessary to process your samples.
Reagents must be defrosted 30 minutes before you arrive, so it is important to keep us updated about any gchanges that happen during the day of the experiment. For example, if there is any delay, there is a change in number of samples, you have to cancel your booking or you know that you will arrive earlier than expected.
We accept samples for our 10x Genomics library preparation primarily from CRUK CI researchers and Institutes who are part of the NovaSeq collaboration. We also try to accommodate experiments from other Cambridge University departments whenever possible. Unfortunately, we do not have the capacity to process samples for commercial companies and researchers from outside Cambridge at this present time.
The submission process if done entirely online and may require a few different accounts to be set up, as this can take some time, we recommend you do it in advance. To be able to submit samples to CRUK CI Genomics Core you will need to:
Usually, one week in advance is enough for us to accommodate your experiment as we keep a small number of reagents in stock. However, larger experiments (requiring more reagents) need to be discussed and booked as soon as possible to give us enough time to place an order. Typically, 10x Genomics deliveries usually take around 1-2 weeks to arrive but some unexpected circumstances can always happen so please do get in touch as soon as possible to avoid disappointment.
We also reserve the right to refuse accepting samples that were not agreed upon in advance or if our schedule is too busy to accommodate more samples in a certain time frame. Although the latter is very unlikely, we do have periods of intensified submissions, including before Christmas and at the end of financial year. If we ever feel that the quality of our service would suffer, it would be necessary to stop taking new bookings.
For example: I lost all my cells during processing, test subject didn't survive or cell line looked very unhealthy before delivery.
We understand that experiments don't always go according to plan, and sometimes submission won't be possible due to circumstances beyond your control. Mostly we can accommodate last minute changes (bringing samples later, more or less samples within reason), however please remember to inform us about complete cancellations as soon as it is possible, especially noting that reagents are being taken out of the freezer 30 minutes before planned arrival.
Unfortunately, we cannot let external users into our lab, so please stick to our core hours when a member of our lab will be able to process your samples for you (see When can I submit my samples?).
CRUK CI researchers who might need to run samples in the night or during the weekends can discuss such experiments with the Core Facility Head. Either come to room 026 or send a ticket to our helpdesk to explore this option.
If you have access to Chromium equipment and would like to submit your libraries for sequencing please refer to our Next Generation Sequencing Submission guide.
The cost of library preparation is around £1200 per sample (which includes reagents and labour) but exact price depends on the source of funding of your grant. Please get in touch with our team to find out your exact costing.
Sequencing is not included in the library preparation service and it will vary depending on the scale of the experiment (experimental design, number of samples, number of cells being captured, sequencing depth etc). We recommend discussing this matter with your bioinformatician. Once these parameters have been decided please contact our sequencing team to determine the cost of sequencing.
| Workflow | Kit used | Sample Type | Volume (µl) | Total Number of Cells loaded |
|---|---|---|---|---|
| 10x Single Cell GEX v3.0 (available only to the end of 2020) | sc 3'mRNA v3 | Cells/Nuclei | 47 | 800 - 16,000 |
| 10x Single Cell GEX v3.1 (NextGEM) | sc 3'mRNA v3.1 | Cells/Nuclei | 43 | 800 - 16,000 |
| 10x Single Cell V(D)J v1.0 | sc 5'mRNA | Cells/Nuclei | 32 | 870 - 17,400 |
| 10x Single Cell ATAC v1.1 (NextGEM) | sc ATAC | Nuclei | 5 | 775 - 15,400 |
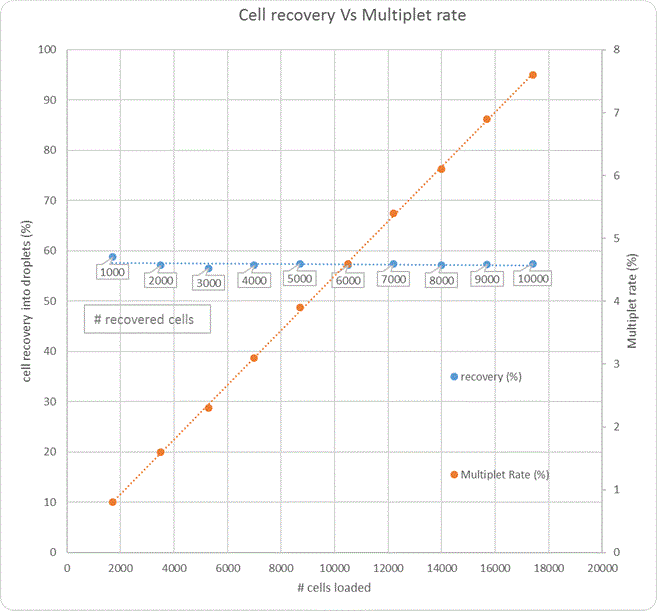
There is no optimal cell concentration when submitting for 10x Genomics experiments. It varies due to cell type, experimental design etc, at least 40% of cells loaded into the chip will be lost but this percentage might be higher in some cell types.
The number of cells that should be loaded into the chip depends on many questions such as:
10x Genomics recommends using Countess II Automated Cell Counter (Thermo Fisher), Scepter Handheld Automated Cell Counter (Millipore) or just a classic manual Hemocytometer. In reality it doesn't matter what method you use as long as you are comfortable with it, it's accurate and doesn't take too long. Please remember that accurate cell counting is critical to the success of the experiment. For additional information on obtaining accurate cell counts please see the following guide.
Unfortunately, we do not perform cell counting or a viability check in our lab, so we rely on you to be confident about the quality and quantity of the cells you give us. As speed is critical in 10x Genomics experiments we load cells into the chip immediately after receiving them from you. We also accept many single cell samples throughout the day, so in order for us to process all of them quickly and provide the best quality service we cannot add any additional steps or delays.
The 10x Genomics website is full of useful protocols, webinars and tips about sample preparation. As every tissue type will behave differently, optimisation of your dissociation method is key to a successful experiment (for example specific enzymes and incubation times may vary). Please follow these general guidelines for preparing optimal single-cell suspensions and use other available resources on the 10x Genomics website, as well as other publications.
It is also possible to use commercially available solutions, for example Miltenyi Micro Beads.
Generally, the cell viability should be >80% and between 1000 and 16,000 cells should be submitted per sample.
It is recommended to run samples in the Chromium within 30 minutes after preparation, as extended time on ice might affect transcriptome. Different cell types will react differently to this delay so it could cause bias towards certain types in samples with multiple cell types.
This is another reason why we do not accept samples prepared outside of Cambridge University.
To prepare single cells for Chromium Single Cell applications, it is recommended to use 1x PBS (calcium and magnesium free) containing 0.04% weight/volume BSA (400µg/ml) for washing and resuspension (BSA concentration can be increased up to 2% to maintain cell health with little to no adverse downstream effects). For primary cells, stem cells, and other sensitive cell types PBS with BSA can be replaced with most common cell culture buffers (DPBS, HBSS) or media (EMEM, DMEM, IMEM, RPMI, Ham's F12, DMEM/F12) + 10% FBS or M199 with minimal reduction in performance. We recommend using anything that is required for keeping your cells viable and intact, and that will prevent aggregation.
However, buffers and media should not contain excessive amounts of EDTA (>0.1mM) or magnesium (>3mM) as these will inhibit the reverse transcription reaction. Any surfactants (Tween-20, etc) should also be avoided as they may interfere with GEM generation.
Please see Cell Preparation Guide for further info.
The maximum cell size tested in-house by 10x Genomics is 30µm, however the theoretical limit is the width of the microfluidic channel, which is ~50-60µm.
What really matters is the actual size of the cells once in suspension. Most adherent cells that are >30µm (e.g. fibroblasts, neuronal cells) will shrink once in suspension and there are experiments where such samples were successfully run. Using the Countess Automated Cell Counter may help here since it provides a distribution of cell sizes that are in suspension.
If you are worried that your cells are too big to be run with the 10x Genomics assay, there is always the possibility of isolating the nuclei (see this demonstrated protocol).
It's also worth mentioning that no differential recovery for small vs large cells was seen when running a sample composed of cells of different sizes (for cells that are equal to or smaller than 30µm).
Yes, many of our users sort cells before submitting for 10x 3'/5' workflows; as FACS is considered good for enriching specific cell types based on cell surface markers and/or removing the dead cells and debris. However, the sorting of single nuclei before ATAC workflow is not recommended as it might compromise the nuclear membrane and cause leakage.
Please remember that long sorting will cause huge stress for the cells, it is possible that the cells sorted first will be apoptotic by the time the last cell exits the sorter.
It is also recommended to recount cells after flow sorting as FACS numbers are usually overestimated.
We have had many users successfully using viability dyes in FACS and we do not have any reason to believe that such reagents interfere with the 10x Single Cell workflow.
Many users ask us if we can "see" how many cells were captured after running the chip. Unfortunately, this is not possible, only sequencing of the library and running the data through the Cell Ranger will determine how successful the experiment was (as Cell Ranger gives metrics such as how many cells were captured, were they still intact when we run a chip etc). This is why it is important to be confident about your cell counts and viability (see question: What cell counting method do you recommend?).
However, there is an intermediate cDNA quality control step on the Bioanalyser/Tapestation. This usually allows us to estimate if your sample is worth proceeding with to sequencing.
If the Bioanalyser/Tapestation trace is completely flat (no cDNA) we will get in touch with you about the failure of the sample and try to troubleshoot what went wrong. In such cases, it is unlikely that sequencing would work well or be an effective use of money, so we would usually stop at this point and not carry on with the library preparation.
There might be multiple reasons for low cDNA including low viability, inaccurate cell count or simply biology: your cell type might be not abundant in RNA. In such scenarios, it is your decision if you would like us to stop processing or if you think it's worth sequencing.
In most cases, the cDNA trace is large, presenting a high yield of cDNA. This doesn't tell us how many cells were actually captured or how good the quality of the sample was but it indicates that everything went according to plan and sample is worth sequencing.
The turnaround time in our facility will vary depending on time of the year so it's worth asking when you submit your samples. It usually takes around four weeks from when you give us samples until you receive your data but this time might even double in particularly busy periods.
Our standard workflow is approximately:
Week 1: We accept samples Monday to Thursday. Your sample will be processed as soon as we receive it and then stored as emulsion in -20°C until Friday. On Fridays, we prepare cDNA of all submitted samples and usually libraries are finished.
Week 2: We prepare your library for sequencing, including QC tests, normalise and pooling libraries.
Week 3/4: After that it will depend on the NovaSeq sequencing queue which can be anywhere between two to six weeks. We will ensure to keep you up to date on this.
One 10x Genomics chip has eight wells, which means we can run between one and eight samples simultaneously. Don't worry if you submit only one sample: the rest of chip will be filled with glycerol and you will be only be charged for the one sample.
If you would like to run more than eight samples in one go, please bear mind that there will be a time delay between chips (samples sitting on ice) which might slightly affect gene expression (in NextGem technology each chip stays in Chromium machine for ~20 minutes).
More importantly, if you are thinking about running more than eight samples in one go please let us know in advance as we may not have enough reagents in stock to accommodate such a large scale experiment.
There is a Containment Level 2 laboratory in the CRUK CI building so we are able to process unscreened human tissues. Currently we cannot accept hazard group 3 biological agents such as HIV, HBV or HCV.
If you are planning to submit CL2 samples, please provide following information BEFORE the experiment, as we will not be able to accept samples with unknown or expired REC studies:
Unfortunately, there is no universal answer to this, as there are many factors to think about when making this decision, such as: how many cells are we expecting to capture; what the experimental question we are trying to answer is (looking for rare transcripts or just differential gene expression between conditions); is your sample RNA rich etc. However, the number of reads you will need is correlated to the number of cells submitted (see question: How many cells should I bring you?).
For gene expression minimum sequencing requirement is 20,000 reads per cell; for ATAC it's 25,000 per nuclei; and for Visium it is 50,000 reads per spot on the slide.
From our experience with gene expression experiments, the minimum recommendation does satisfy some users (and their experimental questions) but many users aim to achieve a more optimal depth of 50,000 reads per cell. We also know users who, when looking for very rare genes, try to obtain 150,000 reads per cell; however, these experiments are very expensive, so we don't see them very often.
Example: You run an experiment with five samples, aiming to recover 10,000 cells in each of them and achieve minimal depth (20,000 reads per cell). This would require one billion reads (5 x 20000 x 10000), which is approximately equal to one lane of S1 flowcell on Novaseq.
An additional difficulty when deciding about number of lanes is, how sure are we that the 60% capture rate will be achieved? If it's your first experiment and you are not sure how your cell type is going to behave, we would not recommend sequencing deeply straight away.
If you are unsure what to expect from your single cell experiment (especially when running many samples in parallel) it is always safer to aim for 20,000 reads per cell or even run your pool on one SP lane on NovaSeq (around 400 million reads) as a QC lane. After the initial CellRanger analysis (when sequencing saturation and number of captured cells is known) libraries can be re-sequenced again, when the sample ratio can be adjusted if needed and aiming for a particular depth.
Yes, we provide such a service. However, the preparation of samples and staining cells with different antibodies/hashtags is still your responsibility (here you can find a protocol from Biolegend where the first section "Cell labelling 10x Genomics platforms" has to be performed in your lab). Please clearly inform us which reagents were used for staining (TotalSeq-A, -B, -C etc) during the submission process so we will be able to prepare additional antibody libraries correctly. There is an ~£85 fee for the preparation of each additional CITEseq library, but this cost may vary depending on the funding of your grant so please get in touch to confirm the price. Please also keep in mind that cell hashing is not officially supported by 10x Genomics. This means it is not included in their software and will require a custom pipeline.
Differences between TotalSeq reagents (from Biolegend website):
For multiplexing using lipid-tagged indices for single-cell and single-nucleus RNA sequencing, please follow the MULTIseq paper.
We don't normally recommend using any fixation methods as freshly prepared samples will give the best data. If it is impossible to run samples immediately, then freezing them might be the best option (see the next question). If neither of these options are possible, this demonstrated methanol fixation protocol is available. Although from our experience, its performance is rather poor in most cases. We have heard about some successful attempts of using this DSP fixation method but our experience with this protocol is still very limited.
Please keep in mind that any other types of fixation (such as formaldehydes/aldehydes) are not compatible with the 10x Genomics chemistry as they disrupt the membrane stability.
Our recommendation will always be to process fresh tissue immediately, but if that's not possible there are protocols that allow the use fresh-frozen tissues.
The tissue should be dissociated first, then this single cell suspension can be frozen in a suitable freezing medium (it is recommended to freeze at least one million cells to recover enough post-thaw. For primary cells we recommend this demonstrated protocol for freezing PBMC.
Alternatively, single nuclei can be isolated from frozen material (for example biobank tissues). Here you can find the "Frankenstein" protocol however; this may require optimisation of some steps.
Here are some guides on how to download and work with raw data:
If you are CRUK CI Researcher you can access your data using the data download tool.
External users need to access their group's FTP account for their data. Your PI will have been sent the login credentials when the account was set up.
Each sample index in the Chromium i7 Sample Index Kit is a mix of four different sequences to balance across all four nucleotides, so you see four "sub-samples" for each actual sample. Please follow our sequencing guide for more help.
It is possible to re-submit old libraries more sequencing if needed. The prepared libraries usually generate enough material to repeat sequencing runs many times (with hopefully none or minimal DNA degradation over time).
We can either:
There is no time limit to request re-sequencing of your old libraries, however; our freezers have limited capacity so we cannot guarantee that we will be able to keep them for longer than two years. You can always request to store the finished libraries yourself; if you think you might need them in the future.
No, the Genomics core only provides the raw sequencing data (demultiplexed into separate indexes) and we do not have capacity to run CellRanger reports for our users.
If you are a CRUK CI researcher, please get in touch with the Bioinformatics core. They have single cell pipelines and will be able to help you with your data.
Unfortunately, we don't have any universal advice for external users. It's worth finding a bioinformatician who will can help with data processing BEFORE submitting samples to our facility (especially as discussing experiment with them might have a huge impact on number of samples/cells/reads needed for the project). If you can't find anyone within your group or institute, there are commercially available consultants or you can learn how to use CellRanger yourself using the tutorials and datasets available on the 10x Genomics website.